85-90 % der HoFH-Patient:innen haben pathogene Varianten im LDLR-Gen1,2


Sehen Sie sich dieses
2-minütige Video zum Mechanismus der HoFH an
HoFH ist mit pathogenen Varianten verschiedener Gene assoziiert, die die Aktivität des LDL-Rezeptors (LDLR) beeinflussen2,3
Der Großteil des LDL wird durch die LDL-Rezeptoren, die sich auf den Zellmembranen der Leberzellen befinden, aus dem Plasma entfernt3
Diese pathogenen Varianten wirken sich auf verschiedene Aspekte der LDLR-Funktion aus und können grob in sechs Kategorien eingeteilt werden:3
 Fehlende Synthese von LDLR oder dem Vorläuferprotein
Fehlende Synthese von LDLR oder dem Vorläuferprotein Defekter LDLR-Transport aus dem endoplasmatischen Retikulum
Defekter LDLR-Transport aus dem endoplasmatischen Retikulum Beeinträchtigte LDL-Bindung
Beeinträchtigte LDL-Bindung Keine LDLR-/LDL-Internalisierung aufgrund defekter Anhäufung in Clathrin-umhüllten Vertiefungen
Keine LDLR-/LDL-Internalisierung aufgrund defekter Anhäufung in Clathrin-umhüllten Vertiefungen Kein LDLR-Recycling
Kein LDLR-Recycling Fehlgeschlagener LDLR-Transport an die Oberfläche der Zellmembran
Fehlgeschlagener LDLR-Transport an die Oberfläche der Zellmembran

Pathogene Varianten in anderen Genen, die die LDLR-Funktion beeinträchtigen, wurden auch bei Patienten mit HoFH identifiziert:3,4:
- APOB: Gen, das Apolipoprotein B kodiert
- LDLR: Gen, das den Low-Density-Lipoprotein-Rezeptor kodiert
- LDLRAP1: Gen, das das Low-Density-Lipoprotein-Rezeptor-Adapterprotein 1 kodiert
- PCSK9: Gen, das das Proproteinkonvertase Subtilisin/Kexin Typ-9-Protein (PCSK9) kodiert
APOB = Apolipoprotein B; LDLR = Low-Density-Lipoprotein-Rezeptor; PCSK9 = Proproteinkonvertase Subtilisin/Kexin Typ 9; LDLRAP1 = Low-Density-Lipoproteinrezeptor-Adapterprotein 1; FH = familiäre Hypercholesterinämie; ARH = autosomal-rezessive Hypercholesterinämie.
Pathogene Varianten verursachen eine Störung des LDL-Signalwegs und reduzieren die Aufnahme von LDL-C aus dem Blut, was zu vorzeitiger Atherosklerose und kardiovaskulären Komplikationen führt1,3-5
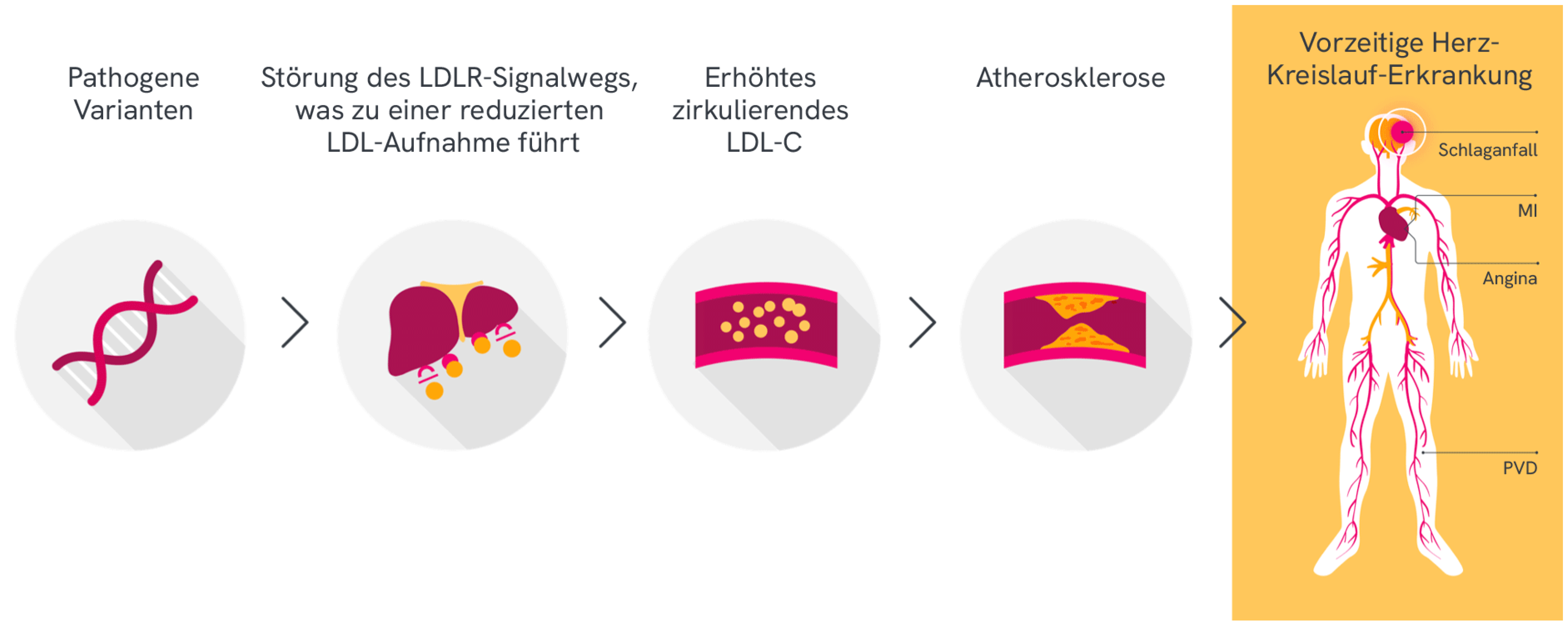
Diese Abbildung gilt für FH generel.
MI = Myokardinfarkt; PVD = periphere Gefäßerkrankung.
Genetische Bestätigung von biallelischen pathogenen/wahrscheinlich pathogenen Varianten auf verschiedenen Chromosomen an den Genen LDLR, APOB, PCSK9 oder LDLRAP1 oder >2 solcher Varianten an verschiedenen Loci.6
LDLR-defiziente pathogene Varianten sind mit geringer oder keiner LDL-Bindung und -Aufnahme assoziiert3
Im Vergleich zu Patient:innen, die einen doppelten LDLR-Defekt und LDLR-Defekt + LDLR-Mangel haben, zeigen sich bei Patient:innen mit doppeltem LDLR-Mangel (null/null):3,6-8
- Höhere LDL-C-Werte
- Vermehrtes Auftreten von kardiovaskulären Erkrankungen
- Schlechtere Prognose
- Reduziertes Ansprechen auf medikamentöse Therapie
Der Großteil des LDL-C wird durch die LDL-Rezeptoren, die sich auf den Zellmembranen der Leberzellen befinden, aus dem Plasma entfernt2

*Das SAFEHEART-Register ist ein landesweites Register, das FH-Patient:innen mit Wohnsitz in Spanien erfasst. Die Patient:innen werden aufgenommen und jedes Jahr einer Nachuntersuchung unterzogen, um relevante Veränderungen in der lipidsenkenden Behandlung und die Entwicklung kardiovaskulärer Ereignisse zu erfassen. Die Grafik zeigt die Überlebensdaten von 34 HoFH-Patienten, die von 2004 bis 2015 aufgenommen wurden.
LITERATUR
1. France M et al. Atherosclerosis. 2016;255:128–139; 2. Cuchel M et al. Eur Heart J. 2014;35:2146–2157; 3. Gidding SS et al. Circulation. 2015;132:2167–2192; 4. Cuchel M et al. Eur Heart J 2023;00:1-15. 5. Nordestgaard BG et al. Eur Heart J 2013; 34:3478-3490;5 6. Bruckert E. Atheroscler Suppl. 2014;15:26–32; 7. Sjouke B et al Eur Heart J. 2015;36:560–565; 8. Alonso R et al. J Clin Lipidol. 2016;10:953–961.