Wenn Sie extrem hohe Werte schlechten Cholesterins (LDL-C) haben, leiden Sie möglicherweise an HoFH.


Was ist HoFH?
HoFH ist eine schwere, lebenslange, extrem seltene Krankheit, die von Ihren Eltern vererbt wird und bedeutet, dass Ihr Körper nicht in der Lage ist, schlechtes Cholesterin (LDL-C) abzubauen. Dies kann zu Plaqueablagerungen in Ihren Arterien führen, die als Atherosklerose bezeichnet werden, und führt oft zu frühen Herzinfarkten, Schlaganfällen und anderen Erkrankungen der Arterien.1
HoFH wird in der Regel mit mehreren Behandlungsansätzen behandelt, unter anderem Änderungen des Lebensstils, Medikamenten und anderen Therapien1.
An HoFH zu leiden bedeutet, dass Sie extrem hohe Werte schlechten Cholesterins (LDL-C) haben, die 5x so hoch oder noch höher sind als der angestrebte Wert (>400 mg/dl bei HoFH gegenüber 70 mg/dl Zielwert für Erwachsene ohne kardiovaskuläres Risiko)1.
Etwa einer von 300.000 Menschen ist von HoFH betroffen. Bei vielen von ihnen wird die Krankheit überhaupt nicht oder erst später im Leben diagnostiziert, was eine wichtige Behandlung verzögert2.
Wie unterscheidet sich HoFH von HeFH?
HoFH und HeFH können ziemlich ähnlich aussehen. Tatsächlich wird die Krankheit bei einer beträchtlichen Anzahl von Menschen zunächst falsch diagnostiziert, da HoFH oft mit HeFH verwechselt wird.
| Homozygous FH (HoFH) | Heterozygous FH (HeFH) |
|---|---|
| Mutierte Gene, die von beiden Elternteilen vererbt werden | Mutiertes Gen, das von einem Elternteil vererbt wird |
| Tritt bei einer von 300.000 Personen auf | Tritt bei einer von 250 Personen auf |
| LDL-C >400 mg/dl (unbehandelt) |
LDL-C-Cholesterin ≥160 mg/dl bei Kindern LDL-C-Cholesterin ≥190 mg/dl bei Erwachsenen |
| Mögliche körperliche Symptome sind z. B. Cholesterinablagerungen in den Augen, Sehnen, Knien, Ellbogen und/oder zwischen Fingern und Zehen. Diese Ablagerungen sind nicht immer vorhanden. | Mögliche körperliche Symptome sind z. B. Cholesterinablagerungen in den Augen, Sehnen, Knien, Ellbogen und/oder zwischen Fingern und Zehen. Diese Ablagerungen sind nicht immer vorhanden. |
| Behandlung zum Zeitpunkt der Diagnose, unabhängig vom Alter, durch einen Spezialisten | Behandlung bereits ab dem 10. Lebensjahr |
Adaptiert von McGowan and Cuchel et al.
Wer erkrankt daran?
HoFH tritt auf, wenn zwei mutierte Kopien von FH-verursachenden Genallelen vererbt werden, und zwar jeweils eine von jedem Elternteil. Diese Eltern haben höchstwahrscheinlich die häufigere heterozygote FH.
Die richtige Diagnose von HoFH kann sich auf ganze Familien auswirken, da auch Eltern und andere Verwandte mit FH diagnostiziert werden. Eine Person mit HoFH kann Geschwister mit HeFH haben.

Was sind die Risiken?
HoFH kann schnell fortschreiten und lebenslange Auswirkungen haben, die zu vorzeitigen Herzinfarkten, Schlaganfällen und anderen kardiovaskulären Ereignissen führen, oft innerhalb der ersten zwei Jahrzehnte.
Eine verpasste Diagnose, Fehldiagnose oder suboptimale Behandlung kann das Fortschreiten der Krankheit begünstigen.
Bei der Geburt
Extrem hohe Werte schlechten Cholesterins (LDL-C bis zu 500 mg/dl bei einem ungeborenen Baby und 700 mg/dl bei einem Neugeborenen)5
Viele Fälle werden im Kindesalter nicht diagnostiziert, wenn das Fortschreiten der Krankheit noch vermeidbar wäre.
Während der Kindheit
Ablagerungen von Cholesterin entwickeln sich in Haut, Sehnen und Blutgefäßen4
Cholesterin sammelt sich in den Herzklappen an, was zu Herzproblemen führt4
Ein Herzinfarkt kann schon vor dem 10. Lebensjahr eintreten6,7
Bei vielen Patienten wird HoFH erst nach Eintreten eines krankheitsbedingten Ereignisses diagnostiziert.
Während der Jugend
Erster Herzinfarkt, Schlaganfall oder anderes herzbedingtes Ereignis4
Cholesterin- und Kalziumablagerungen entwickeln sich weiter4
Fortschreitender Aufbau von Bindegewebe und Entzündungen können zu Herzfehlern führen4
Viele Menschen mit HoFH erhalten erst im Erwachsenenalter eine korrekte Diagnose, was die wichtige Behandlung ihres erhöhten LDL-C-Spiegels verzögert4
Im Erwachsenenalter
Plaqueablagerungen in den Arterien und fortschreitende Erkrankung der Arterien4
Probleme mit den Herzklappen4
Herzversagen, plötzlicher Herzstillstand4
Unbehandelt überleben Menschen mit HoFH selten länger als 30 Jahre4

Anzeichen erkennen

Andere Mitglieder Ihrer Familie, die an familiärer Hypercholesterinämie leiden (dies kann entweder HoFH oder die häufigere HeFH sein)1,3
Extrem hohe Werte schlechten Cholesterins (LDL-C ≥400 mg/dl ohne Behandlung)1
Frühe kardiovaskuläre Ereignisse wie Herzinfarkt oder Schlaganfall4
Sichtbare Anzeichen, wie z. B. Fettansammlungen unter der Haut (sogenannte Xanthome), die vor dem 10. Lebensjahr auftreten
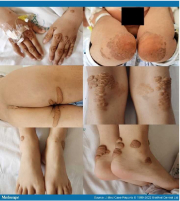
Beispiele von Xanthomen bei Kindern8
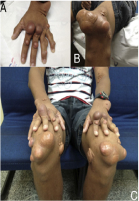
Beispiele von Xanthomen bei Erwachsenen9
Nächste Schritte
Sprechen Sie mit Ihrer Ärztin oder Ihrem Arzt über eine Diagnose. Mit der richtigen frühzeitigen Diagnose können Sie Maßnahmen ergreifen, um Ihr schlechtes Cholesterin (LDL-C) mit einer auf Ihren Zustand zugeschnittenen Therapie zu senken.
Lassen Sie sich sofort von einem Spezialisten (z. B. einem Kardiologen oder Endokrinologen) behandeln.
Lipidolog:innen in Ihrer Nähe findenFinden Sie heraus, ob andere Familienmitglieder vom Risiko einer HoFH-Erkrankung betroffen sind.
LITERATUR
1. Cuchel M et al. Eur Heart J. 2023;00:1–15; 2. Tromp TR et al.The Lancet.. 2022;399:719-728; 3. McGowan MP et al. J Am Heart Assoc. 2019;8:e013225; 4.Cuchel M et al. Eur Heart J. 2014;35:2146–2157. 5. de Gennes JL et al. Arteriosclerosis.1985;5:440 442; 6. Raal FJ et al. Atherosclerosis. 2018;277:483 492; 7. Al-Shaikh AM et al. Cardiol Young. 2002;12:105 112; 8. Wang N et al. J Med Case Reports. 2022;16(290). 9. Rocha VZ et al. J Am Coll Cardiol. 2013;61:2193;